Evaluating Fungal Feature Importance in Predicting Life Expectancy for Cancer Patients
Introduction
Each year, an estimated two million Americans receive a cancer diagnosis. Patient characteristics such as age, gender, and general health status can impact cancer progression and response to treatment modalities like chemotherapy. Nonetheless, a crucial yet often overlooked element that may hold significant sway is the patient’s microbiome. While humans possess approximately 20,000 genes in our DNA, we also harbor a substantial number of microbial genes, ranging from 2 to 20 million throughout our various bodily microbiomes.
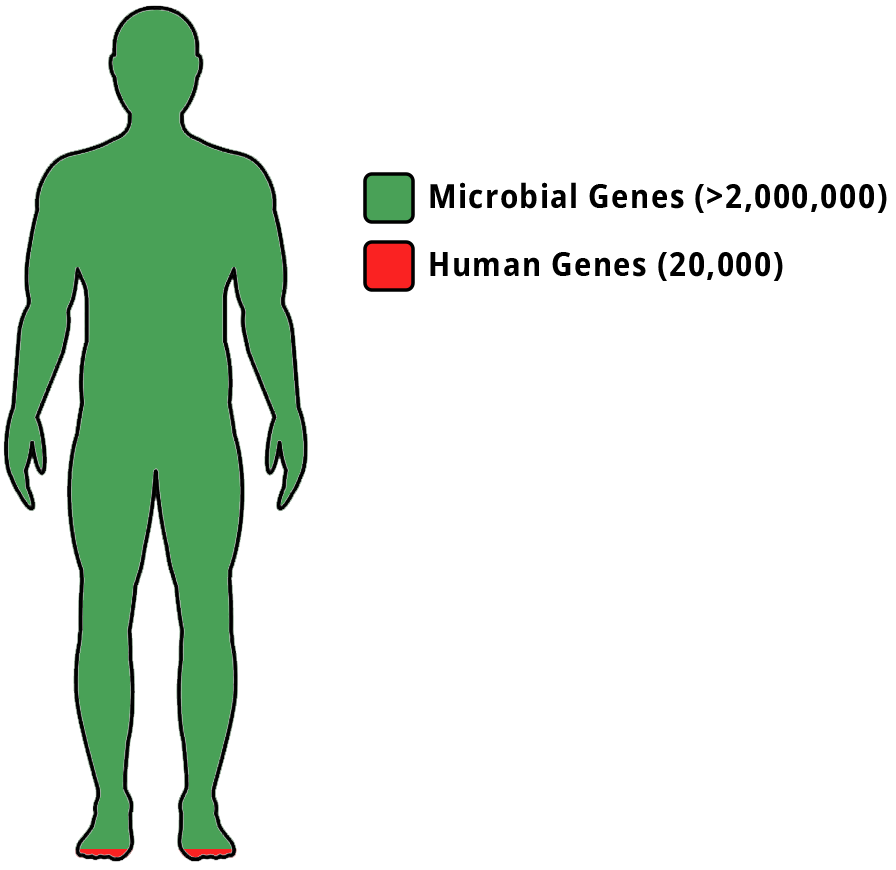
Furthermore, despite a 99.99% DNA similarity between two strangers, their gut microbiomes may only share 10% similarity. In numerous instances, the microbiome composition dictates medication efficacy and disease susceptibility. For example, one study investigated the effectiveness of Cordyceps militaris extract in overcoming carboplatin resistance in ovarian cancer and found that the extract reduced the viability of carboplatin-resistant SKOV-3 cells and induced apoptosis. (Jo et al.)

Consequently, it is plausible that mycobiomes might partly contribute to the differential cancer progression rates observed in some individuals.
Literature Review and Discussion of Prior Work
In the past, researchers have found that bacteria microbes were present in over 1500 tumors spanning seven types of cancer (Nejman et al). The study identified both cancer cells and immune cells as being sites for microbiomes, and that the bacterial composition varied by cancer type. Following this, researchers at the University of California, San Diego re-examined sequencing studies in The Cancer Genome Atlas (TCGA) of 33 types of cancer from treatment-naive patients (a total of 18,116 samples) for microbial reads (Poore et al). They found that they could diagnose cancer type in individuals with stage Ia–IIc cancer and cancers lacking any genomic alterations.
Furthermore, they were able to distinguish between healthy individuals and individuals with multiple cancers solely using microbial signatures. Additionally, a paper published earlier this year also found that multi-kingdom microbiota was effective at diagnosing colorectal cancer (Liu et al).
The study that we based our research off of for this project was the pan-cancer analysis which revealed cancer-type-specific fungal ecologies (Poore et al.). In this study mycobial data sourced from TCGA was used to distinguish between multiple types of cancers tumors. However, this study had intentionally left out metadata from the analysis in order to emphasize mycobial community impact. Our goal for this project was to see if the reintroduction of the metadata could have a positive impact on identifying pathological stage and the days between diagnosis and death for individuals with various cancers.
Methods
Data Cleaning and Preprocessing
First, we obtained 2 feature tables from the original study examining cancer type classification. These feature tables consisted of the final, cleaned TCGA fungal counts and metadata used in the study with 12773 total samples. Next, we preprocessed the metadata using a combination of One Hot Encoding, Ordinal Encoding, Scaler, passthrough, and dropping features. For days to death regression specifically, we filtered outliers greater than 10,000 days to prevent them from skewing the data. We then imputed missing values, which were primarily from the passthrough features, with the column mean and combined the transformed metadata table with the fungal counts table. Lastly, we consolidated certain columns, such as pathologic_stage_label, down to a handful of categorgies effectively eliminating substages. This proved to be useful in both of our models.
Regression
For regression, we used scikit-learn to run lasso ridge regression with 10-folds cross validation on both the fungal data and preprocessed metadata to predict the patients days to death. We tried out other regression models as well including simple linear regression, bayesian regression, and decision tree regression as well. The parameter for our primary model, lasso was an alpha of 0.1
Classification
For classification, we made a gradient boosting classifier with stratified 10-folds cross validation using scikit-learn. For the gradient boost classifier, we used exponential loss, learning rate of 0.1, n-estimators 150, and max depth of 3.
Results and Conclusions
Results
Regression
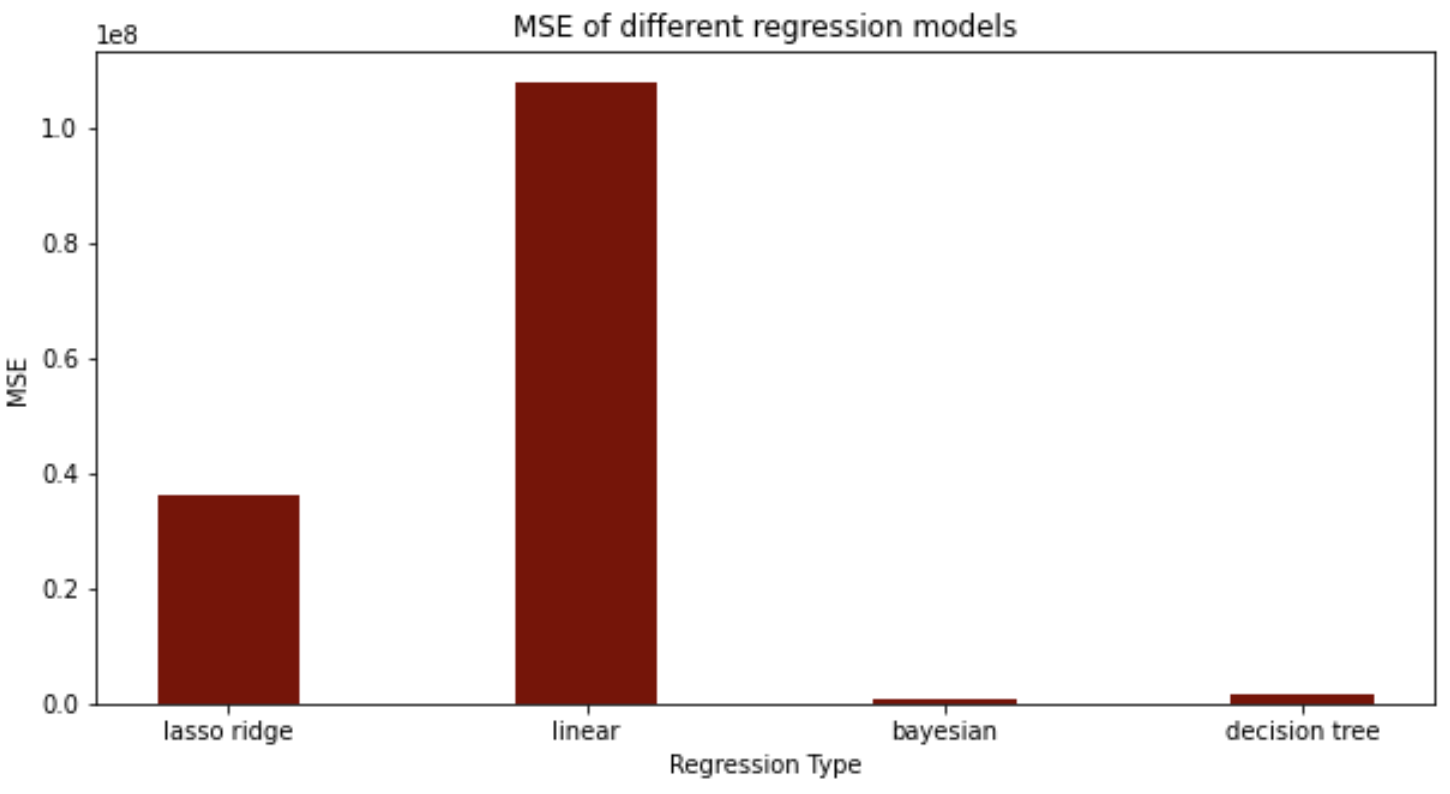
For our regression model, we found that the bayesian and decision tree models performed much better than the lasso ridge regression and linear regression, when comparing the mean squared errors.
Classification
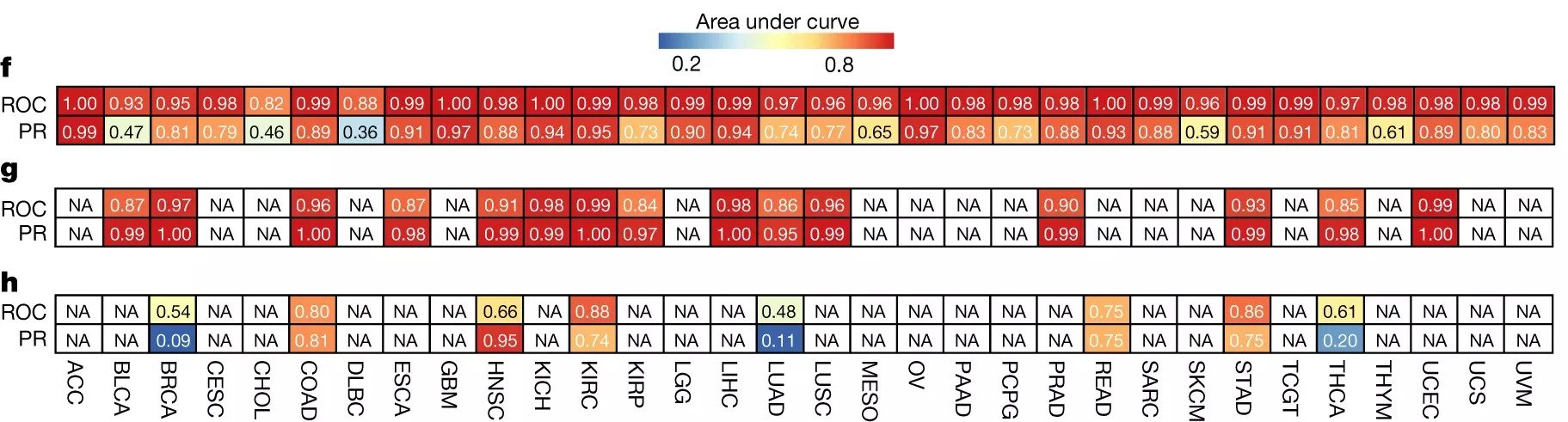
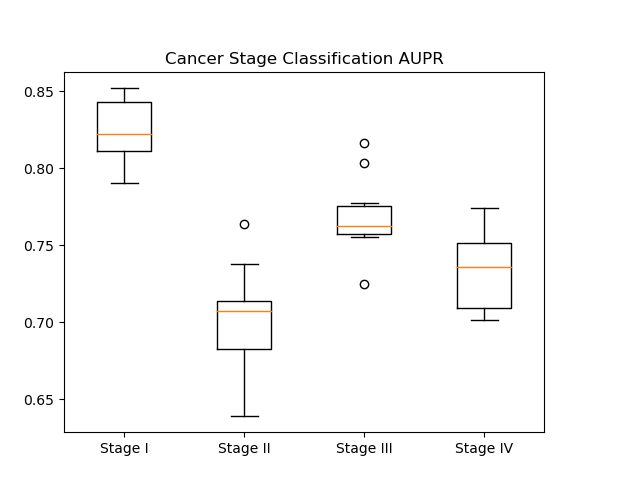
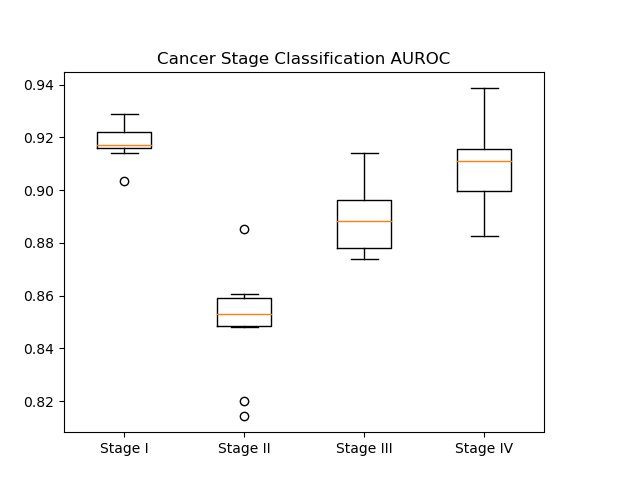
In our results, we generated AUROC and AUPR plots for our classification of cancer stage. AUROC is the area under the receiver operator characteristic curve, which essentially shows our true positive rate. The AUPR plot, or the area under the precision recall curve shows the precision of our classifier.
PCoA
We generated two Principal Coordinate Analysis plots, showing the separation between stages as well as the separation between disease types by a euclidean distance metric. We can see some similar clustering when comparing the two plots, indicating there likely being a relation between the cancer stage and the specific cancer type. This could be a confounding caused by the way that the data was collected, and may be something to explore in further research if there is more data.
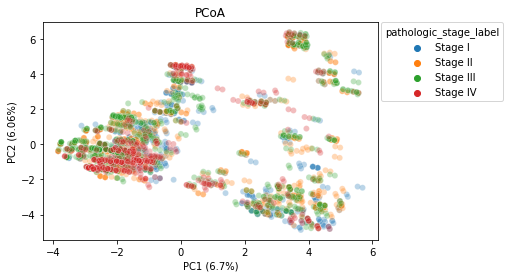
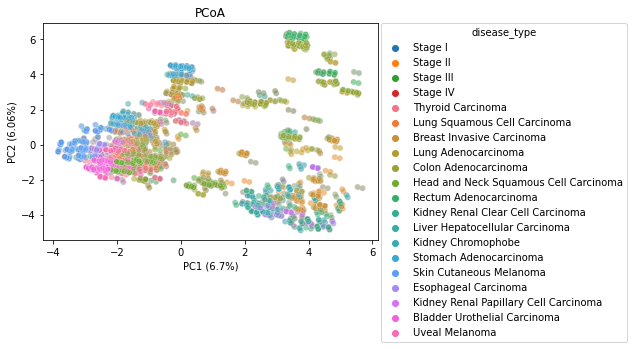
Feature Weights
Finally, we generated bar plots showing the features that had the greatest importance in our classification model. Overall the feature importance for all the stages was relatively similar, but there were some differences especially when comparing between stage I and stage IV.
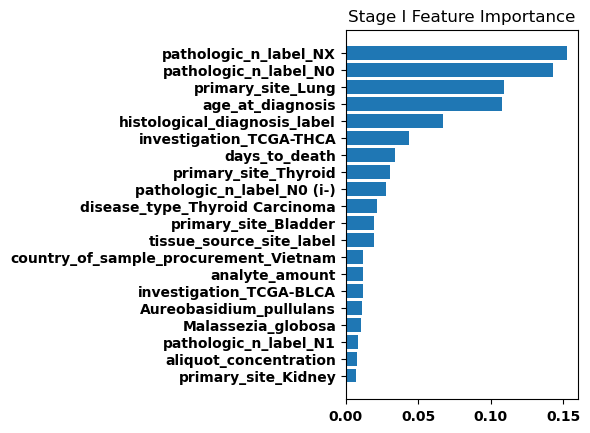
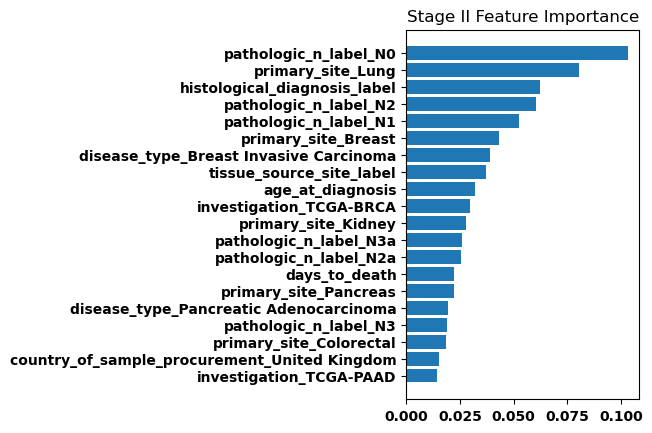
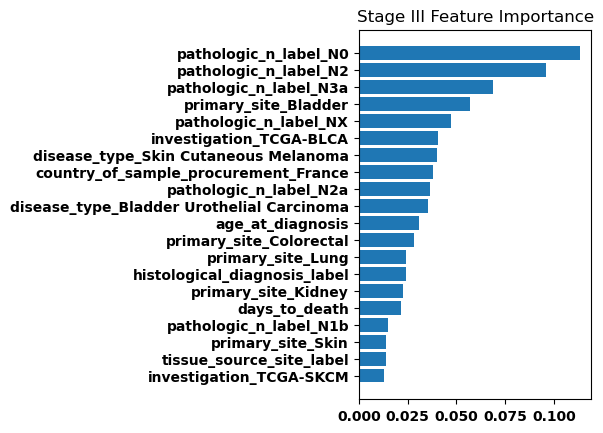
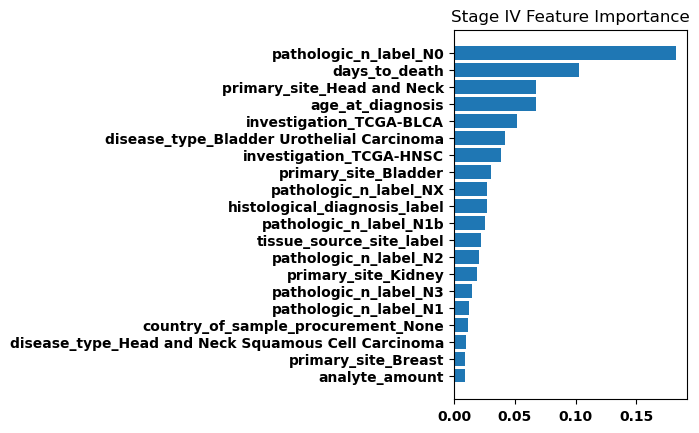
Discussion
Our study successfully achieved a high level of accuracy in classifying the stage of various cancer tumors using a combination of metadata and counts data. Notably, the inclusion of metadata in our model increased model performance compared to the original study. However, the features that our model identified as most important did not include any microbial features. It is possible that the microbial features each had a relatively small effect on the model, making them less significant than the metadata. Future studies may want to investigate methods to boost the impact of microbial features.
Reducing the number of cancer stages to four may have contributed to our model’s performance by reducing the risk of inaccuracy in attempting to classify too many stages.
Additionally, our regression model for predicting days to death was a novel concept not attempted in the original study. Despite utilizing only metadata and counts data, we achieved respectable accuracy levels in our predictions.